Reporte
de caso
Vipoma asociado a dermatomiosistis, tiroiditis de Hashimoto y pancreatitis
autoinmune (síndrome poliglandular tipo 3C)
Vipoma associated
with dermatomyosistis and Hashimoto’s thyroiditis-autoimmune
pancreatitis (polyglandular syndrome
type IIIC)
Hugo
Michael García-Ramos1,Will Omar Delgado-Vera
Médico
asistente. Servicio de Medicina Interna. Hospital Nacional EsSalud Edgardo
Rebagliati Martins, Lima, Perú.
RESUMEN
Mujer de 72 años con diarrea crónica secretora,
disfagia orofaringea, debilidad muscular proximal, compromiso
dérmico y bocio multinodular; con imágenes y estudios auxiliares compatible con
un VIPoma benigno asociado a una dermatomiositis y un
síndrome poliglandular tipo IIIC (tiroiditis de Hashimoto
y pancreatitis autoinmune). Fue tratado con corticoides, inmunosupresores y análogos
de somatostatina con buena evolución clínica. Sería el primer caso reportado en
nuestro país.
Palabras claves: VIPoma, tumor pancreático, dermatomiositis, hipotiroidismo,
tiroiditis de Hashimoto, síndrome poliglandular tipo IIIC.
ABSTRACT
A 72-year-old woman
with chronic secretory diarrhea, oropharyngeal dysphagia, proximal
muscle weakness, dermal involvement and multinodular goiter;
with images and auxiliary studies compatible with a benign VIPoma
associated with dermatomyositis and a polyglandular
syndrome type IIIC (Hashimoto’s thyroiditis and autoimmune pancreatitis). She was treated with
corticosteroids, immunosuppressants
and somatostatin analogues with good clinical
evolution. It would be the first case reported in our country.
Keywords: VIPoma, pancreatic tumor, dermatomyositis, hypothyroidism, Hashimoto’s thyroiditis, polyglandular syndrome type IIIC.
INTRODUCCIÓN
Los tumores neuroendocrinos son
neoplasias provenientes del sistema neuroendocrino difuso, las cuales absorben
y descarboxilan aminas precursoras (APUD, por sus
siglas en inglés).1 Los VIPomas son raros, con una incidencia
de 1 por 10 millones de persona en la población general, lo cual los hace de
difícil diagnóstico.2,3
La dermatomiositis es una miopatía inflamatoria
consistente en debilidad muscular principalmente proximal con o sin dolor y con
alteraciones cutáneas asociadas. La primera asociación entre miositis y
neoplasia fue publicada en 1916.4
Los síndromes poliglandulares
autoinmunes (SPGA)
son desórdenes complejos, heterogéneos, afectando a múltiples órganos, tanto
endocrinos y no endocrinos. Para establecer el diagnóstico debe comprometer al
menos dos o más órganos, y desarrollarse el daño por un componente autoinmune.
En el SPGA tipo 3 hay un compromiso de tiroides asociado con cualquier otro
órgano comprometido de forma autoinmune.5
Reportamos el caso de una VIPoma asociado a un síndrome poliglandular
tipo 3C, siendo el primero en nuestro medio.
PRESENTACION
DEL CASO
Paciente mujer de 72 años, natural de
Trujillo, costa norte de Perú, sin antecedentes patológicos familiares relevantes,
sin hábitos nocivos. Con un tiempo de enfermedad de un año caracterizado por diarrea sin moco y
sin sangre que no cedía con el ayuno, con una frecuencia de 6 a 8 cámaras al
día, de gran volumen (aproximadamente un litro por día), asociado a disfagia a
sólidos que progresó a líquidos. Un mes antes del ingreso, se agregó debilidad
proximal simétrica progresiva de los miembros superiores e inferiores; y, baja de
peso de 10 kilos, motivo por el cual acudió por el servicio de emergencia.
Al ingreso, los signos vitales se encontraban
estables; y, al examen físico, se evidenció eritema periorbitario
violáceos y a nivel del abdomen, dorso y extremidades máculas eritematosas poiquilodérmicas. En el cuello se palpaba un bocio
multinodular difuso y en el examen neurológico impresionaba bradilalia y la
fuerza muscular disminuida a predomino proximal a nivel de las cinturas
escapular y pélvica. El examen restante no fue significativo.
En los exámenes de la hemática sanguínea
y la bioquímica: Hemoglobina 13,2 g/dL, VCM 85,8 fl, HCM 28,9, leucocitos 4 000/mm3, plaquetas 346/mm3,
creatinina 0,39 mg/dL, Na 140,4
mEq/L, K 3,00 mEq/L, DHL 2486, (TGO (AST) 311 U/L, TGP (ALT) 289 U/L, fosfatasa
alcalina 1 770 U/L, GGTP 460 U/L. Bilirrubinas y amilasa dentro de rangos
normales. Creatinin fosfoquinasa
(CPK) 6 555 U/L (VN: 33-211 U/L).
Para el estudio de la diarrea crónica
se solicitó coprocultivo, reacción inflamatoria y parasitológico seriado los
cuales fueron negativos; y, serología para VIH, hepatitis, VDRL, HTLV-1, TORCH,
EBV fueron no reactivas.
Una colonoscopia sin hallazgos
significativos. En las imágenes, la colangioresonancia
evidenció un hígado sin lesiones ni dilatación de la vía biliar intrahepática,
con un colédoco ectasico de 9 mm en su tercio distal,
sin litiasis, con un páncreas de morfología conservada (Figuras 1a- 1c). Ecoendoscopía: parénquima pancreático con estrías hiperecogenicas difusas a predominio de la cabeza, así
mismo se halló una lesión focal de 15 x 10 mm doppler
negativo que ocluía el lumen del colédoco, Wirsung dilatado de 4,5 mm y
adenopatías peripancreaticas de 8 mm.
El resultado de la biopsia mostró un infiltrado inflamatorio linfoplasmocitario y fibrosis periductal,
motivo por el cual se solicitó de inmunoglobulina G4, la cual se encontró en rango
normal. Se concluyó con el diagnóstico de una pancreatitis autoinmune tipo 2.
Finalmente, para completar el estudio de la lesión pancreática, se solicitó cromogranina A: 303,3 ng/ml (VN: 0-100 ng/ml) y péptido
intestinal vasoactivo: 85 pg/ml (VN 0-35 pg/ml) haciéndose el diagnóstico de un VIPoma
pancreático. Así mismo, en la tomografía contrastada de estadiaje no
se evidenció lesiones en otros órganos (Figuras 2a-2d).
Perfil
tiroideo y hormonal


Figura
1.
ColangioRMN. Sin lesiones significativas.
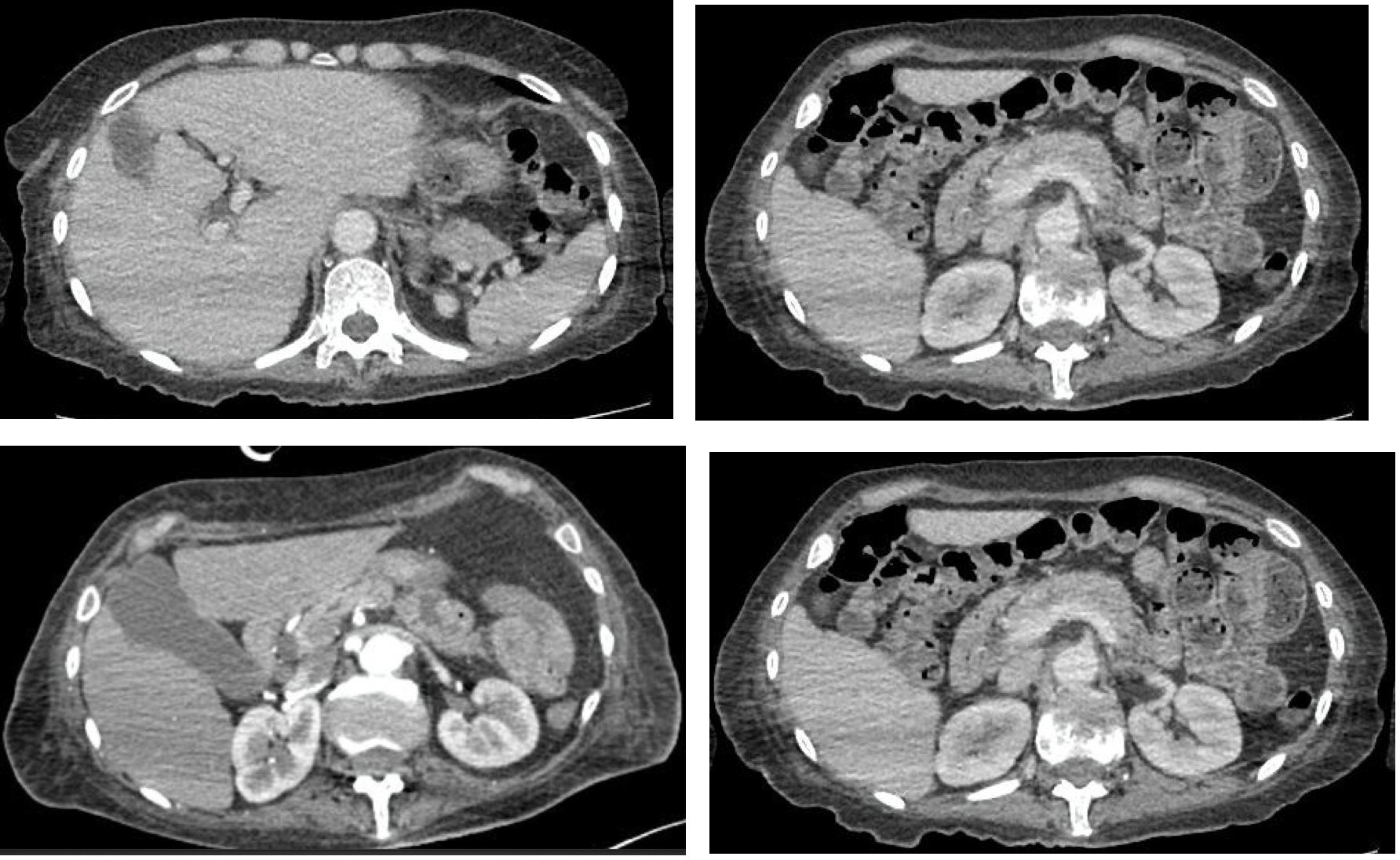
Figura 2a-2d. TAC abdomino-pélvica con contraste. Ausencia de metastásis.
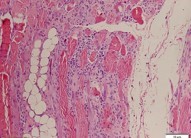
Figura 3. Biopsia de músculo con atrofia e infiltrado inflamatorio
consistente con miositis inflamatoria.
Para el estudio de la debilidad
muscular proximal se realizó una electromiografía la cual reveló un patrón con un
potencial de amplitud muscular disminuido en todos los nervios, compatible con
una miopatía inflamatoria y creatina fosfokinasa
(CPK) muy elevada, con anticuerpos antinucleares (ANA) en una dilución de 1/5120
con patrón nuclear y moteado fino. La biopsia muscular reportó un infiltrado inflamatorio
crónico perivascular y perimisial, así como atrofia perifascicular (Figura 3). Con estos hallazgos sumados a la
disfagia orofaringea progresiva y la afectación de
piel previamente descrita se concluyó el diagnóstico de dermatomiositis.
Finalmente, dentro del estudio del eje
hormonal tiroideo se encontró un cuadro de hipotiroidismo con antiTPO y antiTG elevados (Tabla 1).
Ecografía de tiroides: bocio multinodular TIRADS 3 con biopsia por aspiración aguja
fina cuya descripción era compatible con tiroiditis de Hashimoto. Este último
aunado a la pancreatitis crónica previamente descrita nos hizo el diagnóstico de
un síndrome poliglandular tipo 3 B.
En la evolución, la paciente recibió
pulsos de metilprednisolona por cinco días; sin embargó, la debilidad muscular
progresó y generó un cuadro de insuficiencia respiratorio requiriendo soporte
ventilatorio invasivo en unidad cuidados intensivos (UCI), motivo por el cual
se administró pulsos de ciclofosfamida 0,5 gr/m2 con lo cual hubo mejoría de la
mecánica respiratoria y saliendo de UCI. Al ser una lesión pancreática pequeña,
y por el estado crítico de paciente, no se planteó el manejo quirúrgico y con
terapia de mantenimiento con prednisona a 1 mg/kg más azatriopina
2-3mg/kg/día. Para la diarrea crónica se empleó octreotide
50 mg sc cada 8h y levotiroxina a 1,6 ug/kg/día para la tiroiditis, con buena evolución clínica
saliendo de alta y control por consultorio externo.
DISCUSIÓN
Los VIPomas
son un tipo de tumores neuroendocrinos funcionantes que estimulan la producción
de péptido intestinal vasoactivo (VIP), el cual presenta una estructura similar
a la secretina, actuando mediante sus receptores VIP1 Y VIP2, estimulando la
producción de adenosina monofosfato cíclico (AMPc) por el tracto intestinal y generando
una diarrea tipo secretora abundante mayores de un litro por día, conocida
también como “cólera pancreático”. Otro efecto es generar un hiperaldosteronismo
secundario, manifestado por la secreción de electrolitos causando hipokalemia, hipomagnesemia e hipofosfatemia, entre otros; así
mismo, inhibe la producción acido gástrico generando aclorhidria e incrementa
la glucogenolisis hepática. Fue descrito por primera vez
por Verner Morrison en 1958.6
Schizas et al. en
una revisión sistemática de 65 casos, con una media de edad de 54 años (rango
de 18 a 75 años), fue más frecuente en sexo femenino 53,8%; siendo la cola
páncreas la ubicación más frecuente 53,8%, luego la cabeza 30,8% y cuerpo
15,4%.7 También pueden encontrarse en otras estructuras como bronquios, hígado,
retroperitoneo, tiroides o adrenales y ganglios simpáticos conocidos como neurogénicos.
Solo el 5% está asociado a neoplasias endocrinas múltiples tipo 1 (NEM-1). El
70 a 90 % son malignos y 60 a 50% presentan metástasis al momento del diagnóstico,
siendo el hígado la ubicación más frecuente.7 Ghaferi
et al, en una serie de 35 casos, evidenciaron que el síntoma más común fue la
diarrea secretora en casi el 100%; sin embargo, puede haber otros síntomas como
enrojecimiento facial, distensión abdominal, náuseas o vómitos, letargia y baja
de peso. Para la sospecha diagnóstica se requiere el dosaje de VIP mayor a 75 pg/dl asociado cuadro clínico previamente descrito.6,8,9 En
a las imágenes, la tomografía es muy sensible cuando la tumoración es mayor de 3
cm; y, en caso de menor diámetro, la resonancia magnética y otras pruebas
invasivas como ecoendoscopía son útiles para su
manejo. Finalmente, tenemos al PET- Scan con
trazadores de octreotide, el cual nos ayuda a ver extensión
de enfermedad y seguimiento. Para el estadiaje se usa el sistema propuesto por la
OMS, en 2017, donde se incluye las características histopatológicas y el índice
de proliferación o ki-67.10
Dentro del tratamiento,
en primer lugar,
tenemos a la fluidoterapia y reemplazo
de electrolitos. Según la sociedad norteamericana de tumores neuroendocrinas,
los análogos de somatostatina como el octreotide son la
primera elección para manejo de los síntomas, pueden reducir hasta un 80% la
secreción de VIP y mejorando la diarrea; también se pueden usar loperamida u otros
opiáceos y en casos refractarios se ha visto el beneficio de los antagonistas
de los receptores de serotonina, como ondasetrón. El
abordaje quirúrgico más frecuente fue la pancreatectomía distal seguida de la pancreatoduodenectomía (cirugía de Whipple).
En caso de extensión hepática se puede abordar con resección local cuando son
únicas. Así mismo, la terapia de ablación y embolización serían útiles. En
casos no resecables el uso de everolimus y sunitinib pueden ser empleados con buena respuesta y
mejorando la sobrevida en los pacientes.11,12 Por otro lado, la asociación de
dermatomiositis y cáncer es muy variable con series donde van del 6% al 60%,
generalmente preceden a la neoplasia 5 a 10 años antes; en un estudio de Hill et
al, solo un 3,8% aproximadamente se asoció a cáncer de páncreas. Siendo este
caso poco usual y uno de los primeros reportes de asociación con tumores
neuroendocrino tipo VIPomas.4,13 En todo caso, consideramos que la dermatomiositis
de la paciente estuvo relacionada con el síndrome poliglandular
3C antes que con el VIPoma,
dado que este no era un carcinoma.
Finalmente, en este caso se muestra la
asociación de tiroiditis de Hashimoto y pancreatitis autoinmune lo cual
corresponde a un síndrome poliglandular tipo IIIC
(SPG IIIC), que un trastorno autosómico dominante poligenético
más frecuente en edad adulta, causado por una respuesta inmune humoral y
celular desregulada que genera la producción de citocinas proinflamatorias por los
linfocitos T colaboradores y formación de autoanticuerpos generando
infiltración linfocitaria en órganos específicos, como la tiroides y el páncreas.14-15
En conclusión, dentro del abordaje de
la diarrea crónica secretora, lo tumores neuroendocrinos deben formar parte del
diagnóstico diferencial, siendo de vital importancia en el caso de VIPomas cuando se presenta una diarrea de gran volumen
asociada a trastornos de electrolitos y acido base; así mismo, en todo paciente
con disfagia orofaríngea progresiva asociada a debilidad proximal se debe
plantear la posibilidad de una miopatía inflamatoria, siendo la dermatomiositis
la más común y también recordar que puede preceder a la aparición de una
neoplasia oculta; finalmente, cuando hacemos el diagnóstico de un tiroiditis de
causa autoinmune se debe descartar si hay compromiso en otros ejes hormonales y
buscar compromiso en órganos específicos completando los estudios con la
finalidad de descartar un síndrome poliglandular. Es
el primer reporte en nuestro país de la asociación de estas tres entidades
autoinmunes con un VIPoma,
siendo importante que el médico internista conozca estas, afín
de poder hacer un tratamiento temprano
porque mejora el pronóstico y sobrevida.
REFERENCIAS
BIBLIOGRÁFICAS
1. Öberg K, Eriksson B. Endocrine tumours of the
pancreas. Best Practice of Research
Clinical Gastroenterology. 2005;19(5):753-81.
2. Farina DA, Krogh KM, Boike
JR. Chronic diarrhea secondary to newly diagnosed VIPoma. Case Rep Gastroenterol. 2019;13:225-9.
3. Friesen
SR. Update on the diagnosis and treatment of rare neuroendocrine tumors. Surg Clin.
1987;67:379-93.
4. Hill
CL, Zhang Y, Sigurgeirsson
Ba, Pukkala E, Mellemkjaer
L, Airio A, et al. Frecuency
of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet.
2001;357:96-100.
5. Frommer L, Kahaly
GJ. Autoimmune polyendocrinopathy.
J Clin Endocrinol
Metab. 2019;104(10):4769-4782.